All You Need to Know about FDA Form 483 and Warning Letter


When you are working for a drug manufacturing unit, clinical trial facilities, or a medical device company, you can expect random and routine inspections by the FDA. The FDA regulates food, medicines, vaccines, medical devices, cosmetics, and tobacco products. The riskiest stake a pharmaceutical, biotech, medical devices, food, or cosmetics company can take is to be questionable for its quality practices by the Food and Drug Administration (FDA). When the FDA inspects such facilities, they can either alert the company ahead of time or show up unannounced. Once the inspection is over, the FDA prepares a list of discrepancies and will send an FDA form 483 and, later, a warning letter based on the observation of the inspection.
Depending on the number and severity of the observations made by the inspector, this report could have severe consequences for the pharmaceutical, biotech, medical devices, food, and cosmetics manufacturers inspected. Loss of revenue, time spent on improving compliance processes, and reputational damage are some of the potentially catastrophic effects of a Form 483 and a Warning Letter. Get ahead of Form 483s by getting a Part 11 compliant and validated learning management system and quality management system.
In this article, we will explore Form 483s and Warning Letters and how pharmaceutical, biotech, medical devices, food, and cosmetics companies can ensure they operate within a culture of compliance. Before we proceed, we would like to know that receiving a Warning Letter or FDA 483 does not seem to indicate the end of the product or the company. There are clear procedures to follow to overcome this hurdle.
What is FDA Form 483 Observation?
The FDA sends an FDA Form 483 Observation, also referred to as “inspectional observation” or “Form 483,” to highlight any potential regulatory violations found during a routine inspection. The FDA Form 483 Observation can relate to the company’s facility, equipment, processes, controls, products, employee practices, or records. You might have heard about the KVK-Tech Warning Letter incident?
An FDA Form 483 is issued to company management after an inspection when an investigator(s) has observed any conditions that, in their judgment, may constitute violations of the Food Drug and Cosmetic (FD&C) Act and related Acts. FDA investigators are trained to ensure that each observation noted on the FDA Form 483 is clear, specific, and significant. Observations are made when the investigator’s judgment, conditions, or practices observed would indicate that any food, drug, device, or cosmetic has been adulterated or is being prepared, packed, or held under conditions whereby it may become adulterated or rendered injurious to health.
Historical and Procedural Context of FDA Form 483
The FDA Form 483 is issued at the conclusion of an inspection when the FDA inspector has observed conditions that, in their judgment, may constitute violations of regulatory standards. The form details significant observations and serves as an initial written record of potential non-compliance. As the Wikipedia entry on FDA Form 483 highlights, the content and structure of the form are designed to clearly communicate these observations, providing companies with the opportunity to address issues proactively before they escalate.
What is the Purpose of an FDA Form 483?
The purpose of the FDA Form 483 is to notify the company’s management of objectionable conditions. After an inspection, the FDA Form 483 is presented and discussed with the company’s senior management. Before leaving the facility, the inspector will submit any FDA Form 483 Observations. Upon receipt of the FDA Form 483 Observations, the company has 15 days to respond. Companies are encouraged to respond to the FDA Form 483 in writing with their corrective action plan and implement it expeditiously.
An FDA Form 483 is not a final interpretation of regulation violations; it is a reason for concern and must be urgently addressed. The financial impact of an FDA Form 483 Observation can cost companies several million dollars. The FDA Form 483 Observation can trigger training, redesign, process implementation, and other measures.
What are the Implications of the FDA Form 483 for Agency Enforcement, and What Happens Next?
The FDA Form 483 does not constitute a final Agency determination of whether any condition violates the FD&C Act or any of its relevant regulations. The FDA Form 483 is considered, along with a written report called an Establishment Inspection Report, all evidence or documentation collected on-site, and any responses made by the company. The Agency considers all of this information and then determines what further action is appropriate to protect public health.
Let’s have a look at how the FDA enforcement series flows:
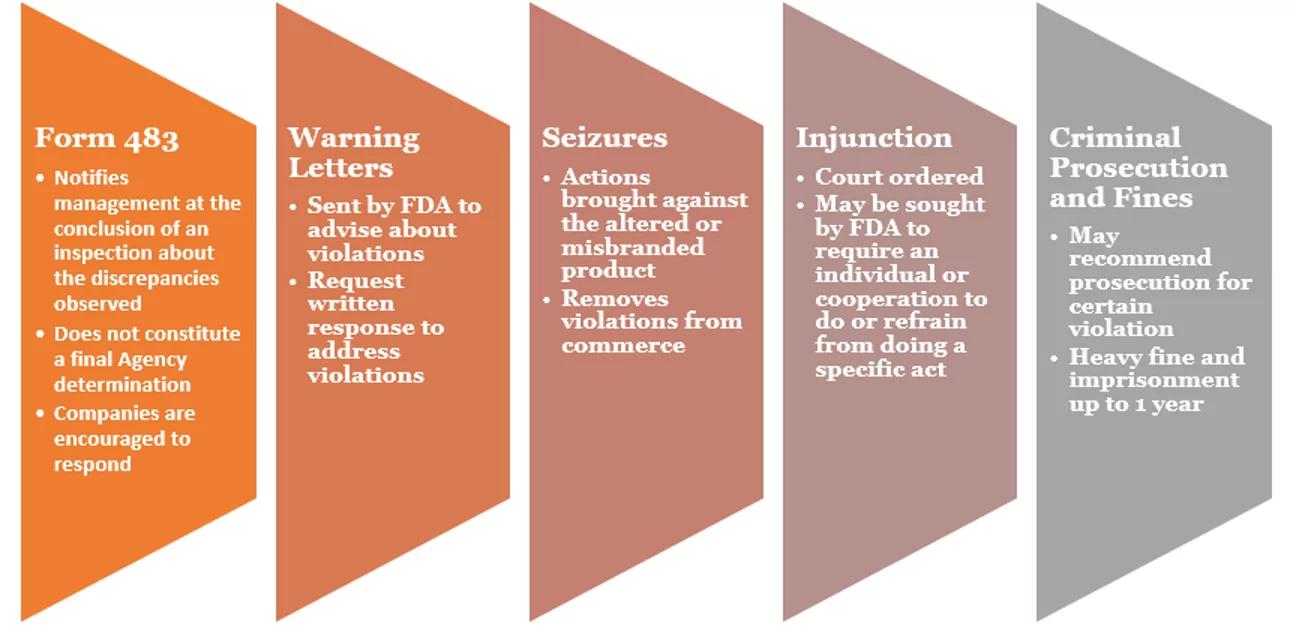
Most Common Causes of a 483 Observation:
- Procedures not fully followed: The pharmaceutical, biotech, medical devices, food, and cosmetics industries must establish standard operating procedures (SOPs) and modify them in a timely manner in order to provide evidence of compliance, quality, and commitment to safety. SOP records must be centrally located and readily available to investigators. When employees aren’t given accurate or current written instructions for their tasks, or if they do not clearly follow written instructions, mistakes and quality problems certainly occur.
- Poor investigations of discrepancies or failures: Deviations from written procedures must be thoroughly investigated and documented. If the company is unable to identify root causes and document them sufficiently, the FDA regards its internal investigations as incomplete. Failure to investigate quality events can result in consequences for a pharmaceutical, biotech, medical devices, food, and cosmetics company.
- Absence of written procedures: As per the FDA observations, written procedures are missing in different pharmaceutical, biotech, medical devices, food, and cosmetics facilities. Companies are not following the rule of thumb rule regarding regulatory industries, i.e., write what you do and do what is written. Many companies struggle to manage and control written procedures, as it can be time-consuming and require additional efforts, especially if the organization is dependent on paper-based processes or if the documentation is scattered between multiple systems in different locales. When deviations or other similar events are not adequately documented or tracked in pharmaceutical, biotech, medical devices, food, and cosmetics manufacturing environments, it’s impossible to pinpoint root causes and take the appropriate corrective and/or preventive actions.
- Data integrity issues: Data integrity has become a common issue in the life science industry. FDA and WHO recently published their data integrity guidelines. This is a frequent issue but very easy to eliminate and doesn’t require working too hard. See how eLeaP can help. FDA found data integrity issues more than 125 times during inspection in the last year, where the data was not secure in the computer system. Digital or physical records must be protected with documented procedures by the companies.
- Cleaning, sanitizing, and maintenance: Cleaning issues in manufacturing facilities are frequently observed during the investigation. Proper cleaning and sanitization of equipment is essential in order to achieve a quality product. FDA can make an observation if the schedules for adjustment, cleaning, and other equipment maintenance have not been established and followed.
- Environmental monitoring: Many companies don’t take environmental monitoring seriously, especially manufacturing facilities. However, the FDA takes it seriously and expects environmental monitoring to be followed, especially for sterile facilities. A proper environmental monitoring program must be prepared and implemented in all classified areas of the facility. Environmental monitoring must be performed at scheduled intervals and should be monitored.
How to Prepare For an FDA Inspection:
Life science organizations should always be inspection-ready. Although there’s no way of controlling what happens during an FDA inspection, there are things a company can do to prepare for it as best they can:
- Close out all CAPAs and ensure that root analysis is performed
- Ensure Data Integrity regulations are followed
- Ensure that all procedures, activities, protocols, etc., are documented.
- Maintain simplified SOPs and update them regularly
- Ensure staff are well-trained on company procedures and have a sound understanding of the procedures followed
- Ensure compliance and quality are followed
- Perform mock inspections regularly to ensure staff know what to expect and that everything is kept up to date.
- Ensure the data is readily available on request.
Preventative Measures and Continuous Improvement
Adopting a proactive stance towards compliance can prevent regulatory issues before they arise. Regular internal audits, continuous training, and a robust quality management system can help identify and rectify potential compliance issues before they attract FDA attention. Companies should view FDA inspections and subsequent documentation not just as a regulatory hurdle but as an opportunity for continuous improvement and strengthening of their quality systems.
What to Do After Receiving an FDA Form 483 Observation
If you have received an FDA Form 483 Observation, it needs to be handled with care. First and foremost, the document must be reviewed with the inspector before he/she leaves the facility. One should have a full understanding of the observations and what needs to be done to correct the issues.
Before the inspector leaves, be sure to:
- Gain an understanding of observations noted and ensure their accuracy
- Understand the broader message the FDA is sending
- Identify and discuss any errors in the observations
- Ask questions
- Demonstrate awareness of applicable regulations
- Consult with legal counsel as necessary
Companies should send the response within 15 days, or the FDA will not be required to consider your response in their decisions for subsequent actions. Once your response is submitted, the FDA will review the Form 483 Observations along with an Establishment Inspection Report and your response. The agency will use this information to determine what further actions to take.
Engaging with FDA Inspectors: A Proactive Approach
Building a collaborative relationship with FDA inspectors during and post-inspection can significantly influence the outcome and subsequent actions. Engaging with inspectors, asking clarifying questions, and discussing the observations can provide valuable insights into their concerns and how best to address them. A proactive dialogue during the exit interview allows companies to understand the context and specifics of each observation, setting the stage for an effective response strategy.
Public Nature of FDA Documentation
Companies in the pharmaceutical, biotech, medical devices, food, and cosmetics industries should be aware of the public aspect of FDA Warning Letters and, in some cases, Form 483s. These documents can affect a company’s reputation and relationships with stakeholders, including customers and competitors. Understanding that these communications may become public underlines the importance of addressing issues promptly and thoroughly to mitigate potential negative impacts on market perception and business operations.
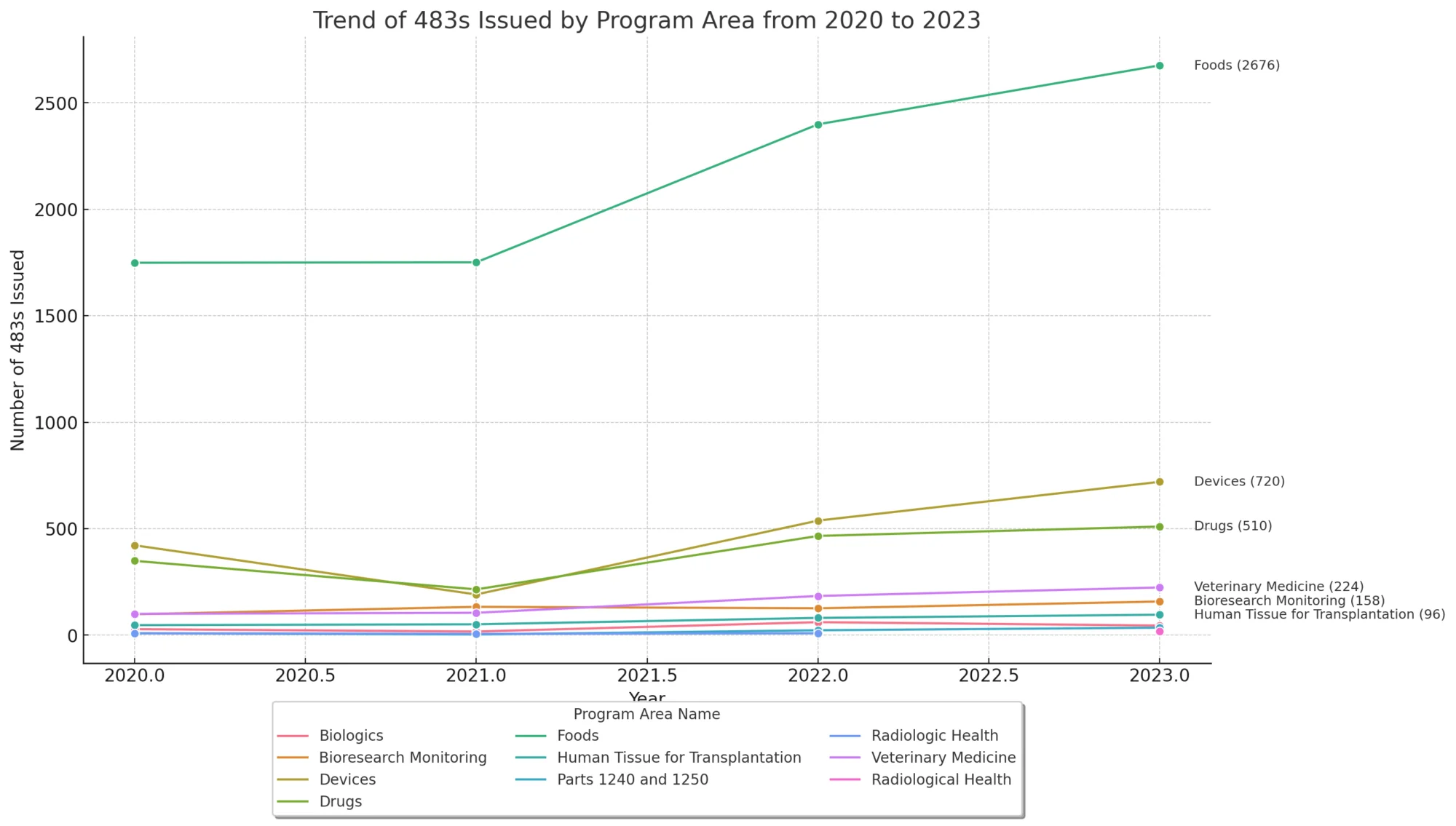
FDA Form 483 Trends
Let’s have a look at the 483 trends from the fiscal year 2020-2023. The below data describes the product and program areas along with the number of the 483 issued:
What is a Warning Letter?
After a Form 483 is issued and the inspector has completed an Establishment Inspection Report, the regulatory agency may issue a Warning Letter. When a serious violation is found regarding the quality of the product, a Warning Letter is issued by the higher level officials after reviewing the observation. The letter makes clear that the company must take action to correct the problem and provides directions and a timeframe for the organization to inform the FDA of its plans for correction.
What is the Difference between the FDA Form 483 and the Warning Letter?
After going through the above details, it’s clear that Form 483 is a notice to inform the company about the potential regulatory violation, while a Warning Letter is a serious escalation of this notice. The company must respond to the FDA in writing within 15 days of receiving Form 483 and a Warning Letter.
Form 483 is not a final determination on regulation violations. These concerns need to be addressed with urgency, while a warning letter is an official deficiency letter issued when quality and compliance are considered unacceptable. Not all 483 observations result in a Warning Letter, but the companies must respond and must be willing to comply with the FDA regulations.
It is essential to understand the critical differences between an FDA Form 483 and a Warning Letter. While a Form 483 outlines observed conditions during an inspection, a Warning Letter is a more serious communication that indicates the FDA’s intention to take enforcement action unless compliance issues are promptly and effectively resolved. The specific information contained in Warning Letters, including CGMP charges and remediation requests, provides clear guidance to companies on the severity of the issues and the steps needed for compliance.
What to Do After Receiving an FDA Warning Letter
When the FDA determines that a company has significant deficiencies with its regulatory policies and issues a Warning Letter, here’s how to respond to the issues raised. The following four steps should be taken:
- Create a Corrective and Preventive action plan
- Immediate acknowledgment of the receipt of the letter
- Develop an action plan to mitigate the issues if one has not been developed. If a plan has been in place, modify it accordingly.
- Determine the most experienced person in the quality group to prepare a written response to the Warning Letter.
Expertise in Navigating FDA Responses
Given the complexities involved in responding to FDA Form 483 and Warning Letters, seeking expert guidance can be invaluable. Specialists in FDA regulatory affairs can assist in interpreting the FDA’s feedback, developing a comprehensive response plan, and implementing corrective actions effectively. This external expertise can help ensure that responses are thorough, compliant, and aligned with FDA expectations, enhancing the probability of a favorable outcome.
Leveraging FDA Form 483 and Warning Letters for Continuous Improvement
Understanding the nuances of FDA Form 483 and Warning Letters is crucial for any FDA-regulated company. These documents not only signify non-compliance but also serve as vital learning tools for the industry. By examining the details and trends in these documents, companies can gain invaluable insights into the FDA’s current focus areas and expectations, aiding in the development of robust quality management systems.
Value of FDA 483s and Warning Letters as Learning Tools
These regulatory documents should not be seen merely as notices of non-compliance but as valuable educational resources. By analyzing Form 483 and Warning Letters, companies can gain insights into common pitfalls and regulatory expectations, helping them to avoid similar issues. These documents offer a practical interpretation of the regulations, illustrating how the FDA applies rules in real-world scenarios. This understanding is instrumental in guiding companies to not only comply with regulations but also to adopt best practices in quality management.
Be Proactive and Prevent Compliance Issues
Companies must always be ready for random FDA inspections. They should keep having the mock-up inspection in order to be ready for the actual one. They should know the most efficient way to avoid Form 483 and know how to respond. If a company has a strong internal audit procedure, this will significantly improve its chances of avoiding a 483 observation. Adhering strictly to quality control regulations and following internal procedures at all times will help staff operate at a consistently high level. Check out a sandbox of eLeaP to allow you to comply with the FDA’s 21 CFR Part 11 regulations.